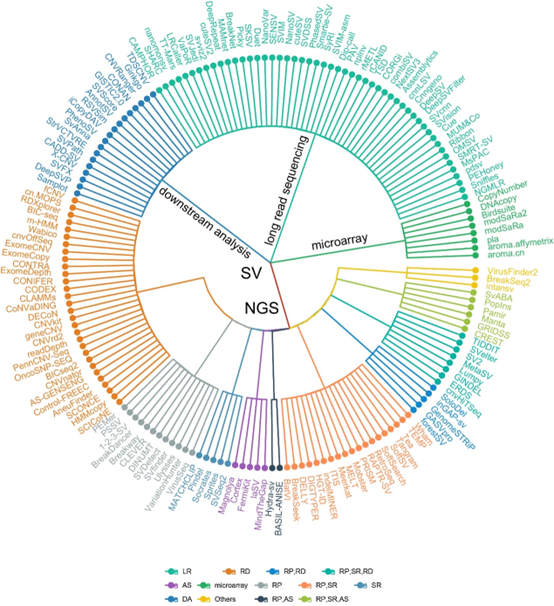
Summary of all tools, including the category of detected SV, name, brief description, download link, reference, input file ,citations, the recommended level, etc.
SV-related tools for CNV
DNAcopy
DNAcopy could process circular binary segmentation (CBS) to transfer intensity measurements into chromosomal regions of equal copy number.
 Potral
Potral
Birdsuite
Birdsuite software has four components (Canary, Birdseed, Birdseye and Fawkes) for processing genotype purpose of CNV and SNP. The uniqueness of Birdsuite lies in that it is capable of integrating CNV and SNP genotype to deriving their mutual effects on phenotypes.
Potral
modSaRa2
modSaRa is a normal mean-based model on a screening and ranking algorithm for copy number variation identification which presented desirable sensitivity with high computational efficiency.
Potral
modSaRa
The modSaRa R package implements modified screening and ranking algorithm (SaRa) and therefore detects CNV from SNP-array.
Potral
pla
PLA can successfully reconstruct the recurrent CNV patterns from raw data and achieve better performance compared with alternative methods under a wide range of scenarios.
Potral
aroma.affymetrix
aroma.affymetrix could process any number of arrays of various chip types (e.g. 10,000s of expression arrays, SNP chips, exon arrays and so on).
Potral
aroma.cn
The R package aroma.cn could use the normalization method called TumorBoost and estimate allele-specific tumor copy numbers from a single pair of tumor-normal genotyping microarrays.
Potral
fCNV
The fCNV software implements unified Hidden Markov Model combining the information of allelic ratios at SNP positions, parental genotypes and depth of coverage.
Potral
cn.MOPS
The cn.MOPS (Copy Number estimation by a Mixture Of PoissonS) software could discover CNV in WGS data with a low false discovery rate (FDR). It takes use of a Bayesian approach and transfers read depth signals into integer copy numbers, noise via its mixture components and Poisson distributions, respectively.
Potral
RDXplorer
RDXplorer is a read depth-based method to detect CNV. Moreover, RDXplorer developed the Event-Wise Testing (EWT) algorithm based on significant testing and has a good sensitivity to CNV which is larger than 1 kb.
Potral
m-HMM
The m-HMM (Mixture-Hidden Markov Model) software based on a hidden Markov model (HMM) takes advantage of Expectation-Maximization (EM) algorithm for solving parameters in the model and thus identify CNV from NGS data.
Potral
Wablco
Wablco is used for correction of GC bias on the basis of multiresolution analysis.
Potral
cnvOffSeq
cnvOffSeq is a novel normalization framework for off-target read depth that is based on local adaptive singular value decomposition (SVD).
Potral
ExomeCNV
ExomeCNV is a statistical method to detect CNV and LOH using depth-of-coverage and B-allele frequencies from mapped short sequence reads.
Potral
ExomeCopy
ExomeCopy is applied to a large chromosome X exome sequencing project identifying a list of large unique CNV.
Potral
CONTRA
CONTRA is a software package that takes standard alignment formats (BAM/SAM) and outputs in variant call format (VCF4.0), for easy integration with other next-generation sequencing analysis packages.
Potral
ExomeDepth
ExomeDepth is a new CNV calling algorithm designed to control for this technical variability.
Potral
CONIFER
CoNIFER can reliably be used to discover disruptive genic CNVs missed by standard approaches and should have broad application in human genetic studies of disease.
Potral
CODEX
CODEX is a normalization and CNV calling procedure for whole exome sequencing data.
Potral
CLAMMs
CLAMMS is highly scalable and suitable for detecting CNVs across the whole allele frequency spectrum.
Potral
CoNVaDING
The CoNVaDING is used to detect simple and compound CNV.
Potral
DECoN
DECoN is a fast, accurate, exon CNV detection tool readily implementable in research and clinical NGS pipelines. It has high sensitivity and specificity and acceptable false discovery rate.
Potral
CNVkit
The CNVkit software is straightforward to use and provides visualizations, detailed reporting of significant features, and export options for integration into existing analysis pipelines.
Potral
geneCNV
The geneCNV software implements a read depth-based method with a hierarchical Bayesian model to conduct the identification of exon-level CNV by exome sequencing data from only a few genes.
Potral
CNVrd2
CNVrd2, an improved version of CNVrd, is also a read depth-based method. CNVrd2 measures copy number at the single gene level or a complex locus using NGS data.
Potral
readDepth
ReadDepth is a parallel R package which can detect these aberrations by measuring the depth of coverage obtained by massively parallel sequencing of the genome.
Potral
PennCNV-Seq
PennCNV-Seq was able to find correct CNVs and can be integrated in existing CNV calling pipelines to report accurately the number of copies in specific genomic regions.
Potral
OncoSNP-SEQ
OncoSNP-SEQ is a statistical approach for the identification of somatic copy number alterations from next-generation sequencing of cancer genomes.
Potral
BIC-seq
BIC-seq can accurately and efficiently identify CNVs via minimizing the Bayesian information criterion.
Potral
BICseq2
BIC-seq2 combines normalization of the data at the nucleotide level and Bayesian information criterion-based segmentation to detect both somatic and germline CNVs accurately.
Potral
CNVnator
CNVnator is an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing.
Potral
MATCHCLIP
MATCHCLIP software identifies the breakpoints and CNV based on WGS or WES data using CIGAR (Concise Idiosyncratic Gapped Alignment Report) strings.
Potral
Magnolya
Magnolya uses a Poisson mixture model for copy number estimation of contigs assembled from sequencing data.
Potral
cnvHiTSeq
cnvHiTSeq is an integrative probabilistic method for CNV discovery and genotyping that jointly analyzes multiple features at the population level.
Potral
ERDS
ERDS accommodates both unique and highly amplified regions of the genome and does so without requiring separate alignments for calling CNVs and other variants.
Potral
iCopyDAV
iCopyDAV is a integrated platform for copy number variations-Detection, annotation and visualization.
Potral
Ginkgo
Ginkgo automatically constructs copy-number profiles of cells from mapped reads and constructs phylogenetic trees of related cells.
Potral
AneuFinder
AneuFinder allows annotation of copy number changes in a fully automated fashion and quantification of CNV heterogeneity between cells.
Potral
CopyNumber
The R package copynumber is a software suite for segmentation of single- and multi-track copy number data using algorithms based on coherent least squares principles.
Potral
SCONCE
SCONCE is a method based on a Hidden Markov Model to analyze read depth data from tumor cells using matched normal cells as negative controls.
Potral
HMMcopy
HMMcopy is a R package for detecting and visualizing CNV
Potral
SCICoNE
SciClone is a computational method that identifies the number and genetic composition of subclones by analyzing the variant allele frequencies of somatic mutations.
Potral
SV-related tools for short read sequencing
SVDetect
SVDetect is a program designed to identify genomic structural variations from paired-end and mate-pair next-generation sequencing data produced by the Illumina GA and ABI SOLiD platforms.
Potral
SVfinder
SVfinder is a tool to detect SV from whole-genome PE or MP sequencing data.
Potral
Ulysses
Ulysses achieves high specificity over the complete spectrum of variants by assessing, in a principled manner, the statistical significance of each possible variant (duplications, deletions, translocations, insertions and inversions) against an explicit model for the generation of experimental noise.
Potral
VariationHunter
VariationHunter is combinatorial algorithms for structural variation detection in high-throughput sequenced genomes.
Potral
Pindel
Pindel is a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads.
Potral
Socrates
Socrates (SOft Clip re-alignment To idEntify Structural variants) is a highly efficient and effective method for detecting genomic rearrangements in tumours that uses only split-read data.
Potral
Sprites
Sprites (SPlit Read re-alIgnment To dEtect Structural variants) is a novel method which finds deletions from sequencing data.
Potral
SVSeq2
SVseq2 enables accurate and efficient SV calling through split-read mapping within focal regions using paired-end reads.
Potral
Cortex
Cortex software, a de novo assembly-based variant detector, takes advantage of colored de Bruijn graphs that provide overlap information within a set of DNA sequences for the de novo assembly.
Potral
FermiKit
FermiKit is a variant calling pipeline for Illumina whole-genome germline data. It de novo assembles short reads and then maps the assembly against a reference genome to call SNPs, short insertions/deletions and structural variations.
Potral
laSV
laSV detects SVs with high accuracy from paired-end high-throughput genomic sequencing data and pinpoints their breakpoints at single base-pair resolution.
Potral
MindTheGap
MindTheGap is used for the integrated detection and assembly of insertion variants from re-sequencing data. Importantly, it is designed to call insertions of any size, whether they are novel or duplicated, homozygous or heterozygous in the donor genome.
Potral
Hydra-sv
Hydra-sv is an algorithm to localize SV breakpoints by paired-end mapping, and a general approach for the genome-wide assembly and interpretation of breakpoint sequences.
Potral
BASIL-ANISE
BASIL-ANISE is the approach for detecting insertion breakpoints and targeted assembly of large insertions from HTS paired data
Potral
BreakSeek
BreakSeek is a novel breakpoint-based algorithm, which can unbiasedly and efficiently detect both homozygous and heterozygous INDELs, ranging from several base pairs to over thousands of base pairs, with accurate breakpoint and heterozygosity rate estimations.
Potral
DELLY
DELLY integrates short insert paired-ends, long-range mate-pairs and split-read alignments to accurately delineate genomic rearrangements at single-nucleotide resolution.
Potral
DIGTYPER
DIGTYPER (Duplication and Inversion GenoTYPER) computes genotype likelihoods for a given inversion or duplication and reports the maximum likelihood genotype.
Potral
indelMINER
indelMINER uses a split-read approach to identify the precise breakpoints for indels of size less than a user specified threshold, and supplements that with a paired-end approach to identify larger variants that are frequently missed with the split-read approach.
Potral
Mobster
Mobster is used to detect non-reference mobile element insertions in next generation sequencing data from both whole genome and whole exome studies.
Potral
PRISM
PRISM (pair-read informed split mapping) is a method that identifies SVs and their precise breakpoints from whole-genome resequencing data.
Potral
RAPTR-SV
RAPTR-SV is a program designed to process previously aligned, Illumina Paired-end whole genome sequence data to identify structural variants such as deletions, insertions and tandem duplications.
Potral
SoftSearch
SoftSearch identifies breakpoints from a small number of soft-clipped bases from split reads and a few discordant read-pairs which on their own would not be sufficient to make an SV call.
Potral
SoftSV
SoftSV is a method for exact breakpoint detection for small and large deletions, inversions, tandem duplications and inter-chromosomal translocations, which relies solely on the mutual alignment of soft-clipped reads within the neighborhood of discordantly mapped paired-end reads.
Potral
Wham
Wham is able to pinpoint SVs in pooled and genotypic data associated with phenotypic variation.
Potral
forestSV
forestSV integrates prior knowledge about the characteristics of structural variants and leads to improved discovery in high-throughput sequencing data. It offers high sensitivity and specificity coupled with the flexibility of a data-driven approach.
Potral
GASVpro
GASVPro is an algorithm combining both paired read and read depth signals into a probabilistic model which can analyze multiple alignments of reads.
Potral
GenomeSTRiP
GenomeSTRiP software could detect DNA segment deletion via the combination between depth of coverage and pair-end mapping method.
Potral
inGAP-sv
inGAP-sv is a novel scheme to identify and visualize structural variation from paired end mapping data.
Potral
SoloDel
SoloDel is a novel computational method that accurately classifies low-frequent somatic deletions from germline ones with or without matched control samples.
Potral
GINDEL
GINDEL is an approach for calling genotypes of both insertions and deletions from sequence reads.
Potral
CREST
CREST (clipping reveals structure) is an algorithm that uses next-generation sequencing reads with partial alignments to a reference genome to directly map structural variations at the nucleotide level of resolution.
Potral
GRIDSS
GRIDSS is a multithreaded structural variant (SV) caller that performs efficient genome-wide break-end assembly prior to variant calling using a novel positional de Bruijn graph-based assembler.
Potral
Lumpy
LUMPY is a novel SV discovery framework that naturally integrates multiple SV signals jointly across multiple samples.
Potral
Manta
Manta is a rapid detection of structural variants and indels for germline and cancer sequencing applications
Potral
MetaSV
MetaSV is an integrated SV caller which leverages multiple orthogonal SV signals for high accuracy and resolution.
Potral
Pamir
Pamir is a new algorithm to efficiently and accurately discover and genotype novel sequence insertions using either single or multiple genome sequencing datasets.
Potral
PopIns
PopIns can discover and characterize non-reference insertions of 100 bp or longer on a population scale.
Potral
SV2
SV2 is a machine-learning algorithm for genotyping deletions and duplications from paired-end sequencing data.
Potral
SvABA
SvABA is an efficient and accurate method for detecting SVs from short-read sequencing data using genome-wide local assembly with low memory and computing requirements.
Potral
SVelter
SVelter is an algorithm that identifies regions of the genome suspected to harbor a complex event and then resolves the structure by iteratively rearranging the local genome structure, in a randomized fashion, with each structure scored against characteristics of the observed sequencing data.
Potral
TIDDIT
TIDDIT utilizes discordant pairs and split reads to detect the genomic location of structural variants, as well as the read depth information for classification and quality assessment of the variants.
Potral
instansv
intansv is an R package for integrative analysis of structural variations.
Potral
BreakSeq2
BreakSeq is the approach for scanning the reads from short-read sequenced genomes against the breakpoint library to accurately identify previously overlooked SVs an approach, for scanning the reads from short-read sequenced genomes against our breakpoint library to accurately identify previously overlooked SVs.
Potral
DeepSVFilter
DeepSVFilter is used for filtering structural variants in short read whole genome sequencing data.
Potral
Cnngeno
Cnngeno is a high-precision deep learning based strategy for the calling of structural variation genotype
Potral
DeepSV
DeepSV is accurate calling of genomic deletions from high-throughput sequencing data using deep convolutional neural network
Potral
AS-GENSENG
AS-GENSENG not only predicts accurate ASCN calls but also improves the accuracy of total copy number calls, owing to its unique ability to exploit information from both total and allele-specific read counts while accounting for various experimental biases in sequence data.
Potral
Control-FREEC
Control-FREEC is a tool for assessing copy number and allelic content using next-generation sequencing data.
Potral
PEMer
PEMer is a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data.
Potral
PSSV
PSSV is a novel pattern-based probabilistic approach for somatic structural variation identification.
Potral
1-2-3-SV
1-2-3-SV detected SV with a high precision and recall rate based on NGS for the capability of detecting four types of SV (deletions, duplications, insertions, and inversions) via benchmark test.
Potral
BreakDancer
BreakDancer sensitively and accurately detected indels ranging from 10 base pairs to 1 megabase pair that are difficult to detect via a single conventional approach.
Potral
Breakway
Breakway is a project dedicated to the identification of genomic breakpoints utilizing freely available tools and custom analytical techniques.
Potral
CLEVER
CLEVER is a clique-enumerating variant finder.
Potral
SV-related tools for long read sequencing
Svision-pro
SVision-pro is a neural-network-based instance segmentation framework that represents genome-to-genome-level sequencing differences visually and discovers SV comparatively between genomes without any prerequisite for inference models.
Potral
Cue
Cue is a deep-learning framework for structural variant discovery and genotyping
Potral
SVcnn
SVcnn is an accurate deep learning-based method for detecting structural variation based on long-read data
Potral
cnnLSV
cnnLSV is applied for detecting structural variants by encoding long-read alignment information and convolutional neural network
Potral
SVDSS
SVDSS is the tool for structural variation discovery in hard-to-call genomic regions using sample-specific strings from accurate long reads
Potral
cuteSV2
cuteSV2 is the tool for regenotyping structural variants through an accurate force-calling method.
Potral
SENSV
SENSV is used for detecting structural variations with precise breakpoints using low-depth WGS data from a single oxford nanopore MinION flowcell
Potral
Duet
Duet is applied for SNP-assisted structural variant calling and phasing using Oxford nanopore sequencing.
Potral
MAMnet
MAMnet is used for detecting and genotyping deletions and insertions based on long reads and a deep learning approach
Potral
SVision
SVision is a deep learning approach to resolve complex structural variants.
Potral
PAV
PAV enables reliable graph-based genotyping from short reads of up to 50,340 SVs, resulting in the identification of 1526 expression quantitative trait loci as well as SV candidates for adaptive selection within the human population.
Potral
SKSV
SKSV is used for ultrafast structural variation detection from circular consensus sequencing reads.
Potral
BreakNet
BreakNet is applied for detecting deletions using long reads and a deep learning approach.
Potral
combiSV
A benchmark of structural variation detection by long reads through a realistic simulated model
Potral
pdsv
The pdsv (PacBio structural variation calling and analysis tools) software developed by Pacific Biosciences is a suite of tools for determining SV for long reads from PacBio sequencing. The pdsv allows CLR reads or HiFi reads as input and map raw BAM files to the reference genome.
Potral
OMSV
OMSV enables accurate and comprehensive identification of large structural variations from nanochannel-based single-molecule optical maps.
Potral
SVIM-asm
SVIM-asm is for structural variant detection from haploid and diploid genome assemblies.
Potral
cuteSV
cuteSV is a sensitive, fast, and scalable long-read-based SV detection approach. cuteSV uses tailored methods to collect the signatures of various types of SVs and employs a clustering-and-refinement method to implement sensitive SV detection.
Potral
NanoVar
NanoVar is used for accurate characterization of patients' genomic structural variants using low-depth nanopore sequencing
Potral
`MUM&Co`
MUM&Co is applied for accurate detection of all SV types through whole-genome alignment.
Potral
MsPAC
MsPAC software combines SNV phasing information and long-read sequencing to discover SV.MsPAC software combines SNV phasing information and long-read sequencing to discover SV. The whole run time of MsPAC could expend 3-8 days on the HG002 dataset from the Genome in A Bottle Consortium (GIAB).
Potral
SyRI
SyRI is used for finding genomic rearrangements and local sequence differences from whole-genome assemblies.
Potral
PhasedSV
Local assembly based SV detection using single-molecule sequencing reads and a phased SNV VCF file.
Potral
SVIM1/2
SVIM is for structural variant identification using mapped long reads.
Potral
Dip-call
A synthetic-diploid benchmark for accurate variant-calling evaluation.
Potral
Smartie-SV
Smartie-SV aligns query contigs against a reference genome and calls SV.
Potral
Picky
Picky comprehensively detects high-resolution structural variants in nanopore long reads.
Potral
NextSV3
NextSV is a meta-caller for structural variants from low-coverage long-read sequencing data.
Potral
CORGi
CORGi is used for detection and visualization of complex structural variants from long reads.
Potral
PEHoney
PEHoney software could detect four types of SV from data produced by the aligners. PEHoney leverages intra-read discordance and soft-clipped tails of long reads to determine the type and length of SV.
Potral
NanoSV
NanoSV is used for mapping and phasing of structural variation in patient genomes using nanopore sequencing.
Potral
SMRT-SV
SMRT-SV could align raw PacBio reads and reference genome, and locally assemble regions for the SV discovery. It currently could detect not only SV containing deletions, duplications, insertions, and inversions but also small indels.
Potral
Assemblytics
Assemblytics is a web analytics tool for the detection of variants from an assembly.
Potral
Sniffles1
Accurate detection of complex structural variations using single-molecule sequencing.
Potral
Sniffles2
Sniffles2 is 11.8 times faster and 29% more accurate than state-of-the-art SV callers across different coverages (5-50×), sequencing technologies (ONT and HiFi) and SV types.
Potral
DeBreak
DeBreak is a computational method for comprehensive and accurate SV discovery.
Potral
SV-related tools for downstream analysis and assisting function
Ribbon
Ribbon is an alignment visualization tool that shows how alignments are positioned within both the reference and read contexts, giving an intuitive view that enables a better understanding of structural variants and the read evidence supporting them.
Potral
TDSCNV
TDSCNV is a tool for inferring haplotypes by implementing CNV or SNP genotype data.
Potral
Ginkgo
Ginkgo automatically constructs copy-number profiles of cells from mapped reads and constructs phylogenetic trees of related cells.
Potral
CNVRanger
CNVRanger is a comprehensive analytic tool for GWAS and difference analysis, which could process variant calling data or genotype data.
Potral
CONAN
CONAN could visualize Manhattan plot depicting significant CNV associated with phenotype and the graph that display all detected CNV in every chromosome.
Potral
GISTIC 2.0
GISTIC has enhanced power and specificity to identify genes targeted by somatic copy-number alterations (SCNAs) that drive cancer growth.
Potral
SVScore
SVScore aggregates per-base single nucleotide polymorphism (SNP) pathogenicity scores across relevant genomic intervals for each SV in a manner that considers variant type, gene features and positional uncertainty.
Potral
AnnotSV
AnnotSV software could fast annotate SV from NGS data with functional and clinical information for identification of disease SV and improve false positive rate for high-quality SV.
Potral
RSVSim
RSVSim is a tool for the simulation of deletions, insertions, inversions, tandem duplications and translocations of various sizes in any genome available as FASTA-file or data package in R.
Potral
iCopyDAV
iCopyDAV is a integrated platform for copy number variations-Detection, annotation and visualization.
Potral
PhenoSV
PhenoSV is the interpretable phenotype-aware model for the prioritization of genes affected by structural variants.
Potral
SvAnna
SvAnna is used for the efficient and accurate pathogenicity prediction of coding and regulatory structural variants in long-read genome sequencing.
Potral
StrVCTVRE
StrVCTVRE is a supervised learning method to predict the pathogenicity of human genome structural variants.
Potral
SVPath
SVPath is an accurate pipeline for predicting the pathogenicity of human exon structural variants.
Potral
CADD-SV
A framework to score the effects of structural variants in health and disease.
Potral
X-CNV
X-CNV is used for genome-wide prediction of the pathogenicity of copy number variations
Potral
SVFX
SVFX is a machine learning framework to quantify the pathogenicity of structural variants.
Potral
DeepSVP
DeepSVP is applied to the integration of genotype and phenotype for structural variant prioritization using deep learning.
Potral
Samplot
Samplot is a platform for structural variant visual validation and automated filtering.
Potral
svviz1/2
svviz is a read viewer for validating structural variants
Potral
SVJedi
SVJedi is used for genotyping structural variations with long reads.
Potral
SVJedi-graph
SVJedi-graph is used for improving the genotyping of close and overlapping structural variants with long reads using a variation graph.
Potral
VaPoR
A recurrence-based approach for validating structural variation using long-read sequencing technology.
Potral
LRCaller
LRCaller is the software for SV assessment based on haplotype-resolved assemblies.
Potral